Healthcare is often measured in appointments, tests, and treatments.
Outside those visits, the same condition is often lived hour by hour—routines, medications, and close watching that do not pause when the appointment ends.
Nowhere is that contrast clearer than in conditions like spinal muscular atrophy (SMA). In Singapore, the media recently drew attention to two children under three living with SMA type 1—the severe infantile form—bringing rare-disease care into public view, if only for a moment.
The real work starts after diagnosis
When Norhaziqah Rosli’s son was diagnosed with SMA, life didn’t revolve around hospital visits. It revolved around everything in between: medications, breathing support, constant monitoring and sleepless nights. Care wasn’t occasional. It became her entire day. (Source: https://cnalifestyle.channelnewsasia.com/women/mother-lost-son-spinal-muscular-atrophy-raising-baby-same-disease-576996)
SMA is a genetic condition caused by a mutation in the SMN1 gene, which is responsible for producing a protein essential for motor neuron survival. When this gene does not work properly, motor neurons in the spinal cord begin to die. Over time, the brain can no longer effectively control muscle movement, leading to progressive muscle weakness.
How SMA is classified
Clinicians group SMA by typical age at onset and severity. The picture is not one-size-fits-all: outcomes range from the most fragile neonatal presentations to mild, adult-onset disease.
Type 0 (congenital SMA) is uncommon. Families may notice reduced fetal movement before delivery; at birth, weakness is profound and respiratory failure is common. Without intensive support, life is often measured in days or weeks.
Type 1 (severe infantile SMA, Werdnig-Hoffmann disease) accounts for roughly six in ten cases. Symptoms appear before six months—limited head control, floppy tone (hypotonia), and struggle with feeding and breathing. Historically, without ventilatory support, many children did not reach their second birthday; modern care and therapies have shifted that trajectory for some families.
Type 2 (intermediate SMA, Dubowitz disease) usually begins between six and eighteen months. Weakness often hits the legs harder than the arms; many children learn to sit but do not walk independently. Respiratory complications remain a major focus. Longitudinal data often cite on the order of seven in ten surviving to age twenty-five, with some living into their thirties and beyond when care is available.
Type 3 (milder childhood-onset SMA, Kugelberg-Welander disease) emerges after the first eighteen months. Leg weakness can make walking difficult, but breathing is often less affected than in earlier-onset types, and life expectancy is frequently close to that of the general population.
Type 4 (adult-onset SMA) is the mildest pattern: symptoms typically start after age twenty-one and progress slowly. Many people stay mobile for years, and lifespan is usually not shortened by SMA itself.
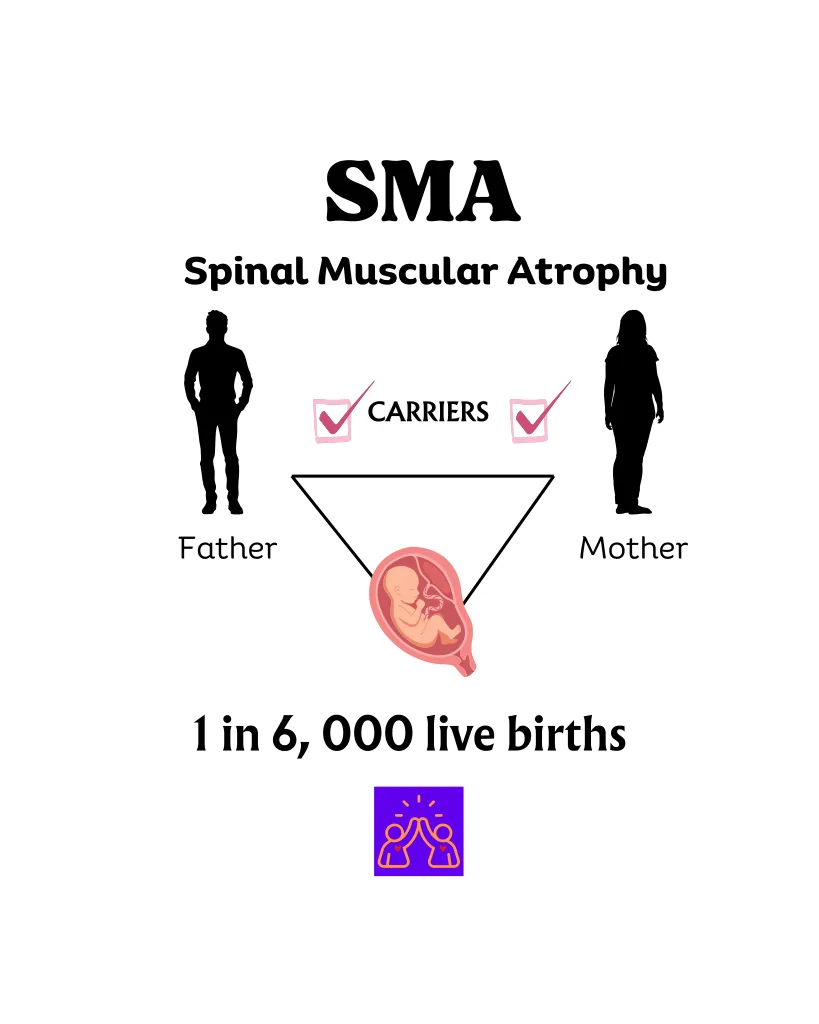
Every person has two copies of the SMN1 gene (one from each parent). A child must inherit two faulty copies to develop SMA. If they inherit only one faulty copy, they are a carrier—healthy, with no symptoms. This is why SMA often appears “suddenly” in families with no known history of the condition. When both parents are carriers, the chances for each pregnancy are as follows:
- 25% chance the child will have SMA.
- 50% chance the child will be a carrier (but not affected).
- 25% chance the child will inherit two healthy genes.
This means that even parents who are completely healthy—and often unaware they are carriers—can still have a child with SMA.
One in every 6,000 babies is born with SMA and it can strike a child at any age. Interestingly, it doesn’t affect the nerves nor intellect (Source: https://www.hopkinsmedicine.org/health/conditions-and-diseases/spinal-muscular-atrophy-sma)
It is a progressive genetic condition that weakens muscles over time, eventually affecting a person’s ability to move, swallow, and breathe. It does not pause between doctor visits. It does not wait for clinical intervention.
Yet most healthcare systems are designed around snapshots: appointments, tests and emergency interventions. What they miss is: Did the patient sleep well? Are they weaker today than yesterday? Is something subtly changing?
The most critical factor in treatment is timing. Early intervention can preserve function. Delayed treatment can mean irreversible decline. This creates a narrow window where action matters most and where delays carry long-term consequences.
When cost becomes the barrier to survival
Today, treatments for SMA can significantly improve quality of life. But they come at extraordinary cost. As one-time gene therapy can exceed $2.4 million or ongoing medication can cost hundreds of thousands annually. For many families, this shifts the challenge from medical to financial.
These costs are largely driven by the complexity of developing treatments for rare diseases, where small patient populations must absorb high research and development investments, as well as the advanced biotechnology required for gene-based therapies. According to the National Organization for Rare Disorders, rare disease treatments often carry significantly higher prices due to limited patient pools and specialised manufacturing processes. (Source: https://rarediseases.org/)
For adult patient Sherry Toh, access to treatment was partial and temporary. After receiving a short course of medication, she experienced clear improvements: daily tasks became easier, and her breathing stabilised. But when funding ran out, treatment stopped. Her condition regressed. The difference between progress and decline was not clinical. It was financial continuity. (Source: https://www.channelnewsasia.com/today/ground-up/living-spinal-muscular-atrophy-patients-pay-millions-drugs-or-face-rapid-decline-inquality-life-4647426)
Technology’s role is not to replace care. It is to support care for spinal muscular atrophy (SMA) and similar genetically inherited conditions.
For many caregivers, the idea of technology in healthcare brings hesitation. Not because they reject help but because they fear more complexity: another app, another system and another thing to manage in an already overloaded day. But the opportunity is not to add more work. It is to remove it.
- To reduce the constant mental calculation.
- To surface what matters without forcing caregivers to actively search for it.
- To create small moments of clarity in an environment defined by uncertainty.
- Not by replacing caregivers but by standing quietly behind them.
This is especially critical in conditions like spinal muscular atrophy (SMA), where change is not sudden; it is gradual and often invisible until it becomes urgent. SMA does not progress in isolated clinical moments. It evolves in the home. And what matters most is not just medical records but daily patterns that are easy to miss:
- Is breathing more effortful today than yesterday?
- Is feeding taking longer than usual?
- Is posture subtly changing over weeks?
- Is fatigue increasing earlier in the day?
- Are movements becoming less frequent or less symmetrical?
- Are recovery times after activity getting longer?
These are not hospital metrics. They are lived signals. And they are often first noticed by caregivers, long before they appear in clinical assessments. Easily dismissed as anecdotal evidence but vital in identifying sudden or new patterns in their development. This is why timing is everything in SMA. Early intervention can preserve function. Delayed recognition can mean irreversible loss. Which means the most important data is not episodic. It is continuous.
And yet, the story of SMA is no longer defined only by decline. It is increasingly a story of progress—of children living longer, of abilities being preserved, and of families gaining support in ways that did not exist before. What was once an invisible struggle is slowly becoming a space of innovation, attention, and hope.
